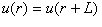In this the final chapter of the thesis perhaps the most interesting results will be presented. Here we will start with a short review of harmonic lattice the-ory together with a brief discussion of how lattice dynamics can be calculated from ab-initio theory. Here special focus will be on the so-called supercell method, since this is the method that has been used through out this thesisto calculate phonons from ab-initio theory. After this brief introduction theresults obtained within the harmonic, or rather quasi harmonic, approxima-tion will be presented (see papers III, IV and VI). The chapter is ends with adiscussion of the anharmonic lattice and a presentation of the self-consistentab-initio lattice dynamical (SCAILD) approach, and the results obtained withthis novel approach (see paper V) will also be discussed.
The Born Oppenheimer approximation
Before discussing the theory of lattice dynamics and the associated calcula-tional methods, it is important to take a closer look at one of the fundamental approximations used in calculating phonons from first principles. This approx-imation is commonly known as the Born Oppenheimer approximation, and itassumes that the electronic response to an atomic displacement is instanta-neous, making it possible to separate the electronic and the ionic subsystems.To convince oneself of the soundness of this approximation one should remember that the typical ionic mass mi is ∼ 105 times bigger than the mass ofan electron me and that the typical kinetic energy of an electron Eke is 103 times bigger than the typical ionic kinetic energy Eki, implying that the ratiobetween the typical velocity of an electron ve and that of an ion vi becomes(ve/vi)=%Ekemi/(Ekime) ∼ 104. Thus from the "perspective of an electron",the ions will always seem to have fixed positions. Hence if U(R) are the de-viations of the ions from their equilibrium positions at a snapshot in time, itis always possible to retain the total energy of the system, at that snapshot,by means of a static electronic structure calculation. Thus, through a seriesof electronic structure calculations, the potential energy of the ionic subsys-tem can be parameterized in terms of ionic deviations. It is general practice toexpress the potential energy in the Hamiltonian of the ionic subsystem, as aTaylor expansion around the equilibrium ionic configuration.


The harmonic lattice
In the harmonic lattice approximation the atomic deviations are assumed to beso small that the potential energy is well described by the second order term in(8.1). This is generally a good approximation, at least at relatively low temper-atures. Later on in this chapter examples of situations will be given in whichthe harmonic approximation fails, such as the high temperature bcc phase ofTi, Zr and Hf. Furthermore, in order to make the notation more transparent,the notation of a monoatomic lattice will be adapted without any loss of gen-erality. The harmonic Hamiltonian in the case of a monoatomic lattice is given by
In the harmonic lattice approximation the atomic deviations are assumed to beso small that the potential energy is well described by the second order term in(8.1). This is generally a good approximation, at least at relatively low temper-atures. Later on in this chapter examples of situations will be given in whichthe harmonic approximation fails, such as the high temperature bcc phase ofTi, Zr and Hf. Furthermore, in order to make the notation more transparent,the notation of a monoatomic lattice will be adapted without any loss of gen-erality. The harmonic Hamiltonian in the case of a monoatomic lattice is given by

In the harmonic approximation, the ionic displacements UR satisfy Bornvon Karman periodic boundary conditions. This means that the displacementscan be expressed as a superposition of plane waves with wavevectors k ∈ 1BZ. Hence the canonical coordinates UR and PR appearing in (8.2), can beexpressed in terms of a new set of canonical coordinates Qk,s andPk,s, i.e

The supercell method
In the previous section it was shown that once the force constant matrix has been calculated and Fourier transformed, the phonon frequencies are easilyaccessed by a simple diagonalization. Fortunately there exists a fairly simpleand straightforward method for calculating from first principles, namely theso-called supercell method. The foundation of the method is provided by theHellman-Feynman theorem, stating that the force FR acting on an atom withspatial coordinate R.
From the above linear relation and the symmetry of the crystal the forceconstant matrix can then be easily calculated. The number of displacementsneeded to retain depends on the symmetry of the crystal. For instance inthe case of the bcc or fcc structure one displacement is sufficient, while in thecase of the hcp structure two displacements are needed.However since, at least in principle, Φij(R)→0 only as R→∞, and sinceonly finite sized supercells can be used, the summation in (8.5) has to be trun-cated, and the dynamical matrix can only be approximately calculated. Fur-thermore, due to the periodic boundary conditions employed in the electronicstructure calculations, the linear relation (8.16) is only true if an infinite sizedsupercell is used. In real life all the periodic images of the displaced atomcontribute in the induction of the forces in the supercell. The correct linear re-lation between force and displacement(s), to be used in a supercell calculation.
Some thermodynamics and the quasi harmonic
approximationIn this section relations between the harmonic phonon spectrum and differentthermodynamic quantities, such as the free energy, internal energy and meansquare atomic deviation, will be derived and briefly discussed. Furthermore ashort presentation of the quasi harmonic approximation will also be given.
The two above expressions for the internal- and free-energy have been used inthe context of the so-called quasi harmonic approximation to calculate Equations of state, Hugoniots and thermal expansions (see papers III, IV and VI).What now remains in this section is a short discussion of the quasi harmonic approximation. This is the most simple approximation dealing with theeffects of anharmonicity in which the anharmonicity related to the terms oorder > 2 in the Taylor expansion (8.1) is neglected, only taking into accounthe anharmonicity related to the force constants dependence upon symmetryconserving strain. The simplicity of this approximation lies in the fact thafor each symmetry conserving strain the lattice dynamics of the system isregarded as being harmonic, permitting the use of the supercell method separately for each symmetry conserving strain. In Fig. 8.2 the phonon density ostates for fcc Au calculated with the supercell method for three different volumes are displayed, as an example of the volume dependence of the phonon spectra.
Thermal expansion
In this section the art of calculating thermal expansion coefficients from firstprinciples will be discussed. This discussion will be based on the work donein papers IV and VI of the these.
Thermal expansion of cubic metals
The calculation of the thermal expansion of elements with cubic symmetry isvery straightforward when done in the quasi harmonic approximation. Firstthe phonon and electron density of states together with static lattice energy iscalculated for a number of volumes around the T = 0K equilibrium volume.Then using Eq. (8.28-8.30) the total free energy is calculated for the differentvolumes at constant temperature and fitted to some EOS.
Thermal expansion of hexagonal metals
To calculate the thermal expansion of hexagonal metals, the free energy andstatic lattice energy have to be parameterized with respect to two degrees offreedom. The most general second order parameterization of the free latticeenergy, allowing only symmetry conserving strains, can be expressed with thesix dimensional strain vector ¯ ε =(ε1, ε1, ε3,0,0,0) and the elastic constants.Using this strain vector together with the definitions given in chapter 5, thestatic lattice energy U.
ORLANING COLMENARES
C.I.V.- 18.991.089
BIBLIOGRAFIA:
web.mac.com/petros...2/.../urn_nbn_se_uu_diva-8198-1__fulltext.pdf
Connect to the next generation of MSN Messenger Get it now!












 n=0,1,2…..
n=0,1,2…..

 This energy may be considered either as the time-averaged energy for a particular atom, or it can be thought of as the average energy of all the atoms in the assembly at any instant in time.
This energy may be considered either as the time-averaged energy for a particular atom, or it can be thought of as the average energy of all the atoms in the assembly at any instant in time.